[Tham khảo:
+ “Đọc khí máu động mạch”- BS Dương Tấn Khánh.
+ “Khí máu động mạch”- ThS BS Bùi Xuân Phúc: https://www.slideshare.net/PhoToRapPhieu/buoi-1-khi-mau-dm
+ “Acid – Base, Fluid and Electrolytes Made Ridiculously Simple”, 3rd edition- Richard A. Preston
+Serie “Renal Tubular Acidosis” của David Li]
I. Các cơ chế duy trì pH cơ thể:
- Hô hấp: thông qua CO2 (bù trừ nhanh).
- Thận: thông qua HCO3- (bù trừ chậm hơn nhưng mạnh).
- Hệ đệm (buffer system): Hb, bicarbonate, … (không khảo sát vì không nhiều ứng dụng trong đọc khí máu)
Phương trình thể hiện sự bù trừ acid-base:

Nguyên lí Le Chartelier, nếu CO2 giảm thì phương trình sẽ chuyển dịch theo chiều nghịch, tức giảm HCO3- => giảm H+ => pH tăng và ngược lại. Đây chính là cơ sở bù trừ trong cơ thể (thông qua phổi và thận) đáp ứng với pH lệch khỏi giá trị bình thường.
II. Thứ tự đọc một kết quả khí máu động mạch:
Lần lượt trả lời các câu hỏi sau:
1/ Bệnh nhân nhiễm toan hay kiềm máu? (pH> 7,45 => kiềm máu, pH< 7,35 => toan máu).
2/ Rối loạn toan kiềm này NGUYÊN PHÁT là rối loạn hô hấp hay chuyển hóa? (kiềm hô hấp/ kiềm chuyển hóa/ toan hô hấp/ toan chuyển hóa?) Lưu ý rằng sẽ có một thông số (PaCO2 hoặc HCO3-) giải thích được tình trạng toan/ kiềm đã được xác định ở câu 1, thông số còn lại liên quan đến cơ chế bù trừ.
3/ Tình trạng bù trừ có hợp lí hay không? Nếu không thì bệnh nhân đang có một rối loạn khác đi kèm. Hô hấp sẽ được bù trừ bằng chuyển hóa và ngược lại (ví dụ nếu bệnh nhân bị toan chuyển hóa thì hô hấp sẽ bù trừ bằng cách “kiềm” đi, tức giảm pCO2), trong đó việc hô hấp bù trừ cho chuyển hóa diễn ra nhanh (bằng cách tăng/ giảm thông khí) trong khi việc chuyển hóa bù trừ cho hô hấp diễn ra chậm hơn (liên quan cơ chế giữ/ thải HCO3- ở thận).
4/ Chẩn đoán (chẳng hạn toan hô hấp kèm kiềm chuyển hóa)? Dựa vào lâm sàng thì nghĩ tới nguyên nhân gì?
III. Lý thuyết về 4 loại rối loạn toan/ kiềm, hô hấp/ chuyển hóa:
Có thể nhớ nôm na rằng rối loạn hô hấp đơn thuần liên quan đến CO2, tức vấn đề nằm ở sự tăng/ giảm thông khí. Trong khi đó, rối loạn chuyển hóa đơn thuần liên quan đến HCO3-, tức vấn đề nằm ở chuyển hóa các chất trong cơ thể và/ hoặc sự đệm của thận.
1/ Nhiễm kiềm hô hấp:
Nhiễm kiềm hô hấp (pH> 7,45, pCO2 < 35 mmHg) là quá trình giảm trực tiếp CO2 máu, từ đó tăng pH máu. Nguyên nhân thường gặp là do tăng thông khí (hyperventilation), rối loạn thần kinh, các bệnh phổi (suy hô hấp, thuyên tắc phổi, …), thiếu O2 máu nên bù trừ bằng thông khí, …
2/ Nhiễm toan hô hấp:
Nhiễm toan hô hấp (pH <7,35, pCO2 > 45 mmHg) là quá trình tăng trực tiếp CO2 máu, từ đó giảm pH máu.
Nguyên nhân thường gặp:
- Giảm thông khí có nguồn gốc trung ương: bệnh lý của não; tủy, hội chứng ngưng thở khi ngủ (obstructive sleep apnea- OSA).
- Giảm thông khí có nguồn gốc ngoại biên: hội chứng Guillain Barré, nhược cơ hô hấp, gù vẹo cột sống, …
- Tắc nghẽn đường thở.
- Thông khí nhân tạo: cài đặt thông số không phù hợp, chiến lược bảo vệ phổi chấp nhận tăng CO2.
3/ Nhiễm kiềm chuyển hóa:
Nhiễm kiềm chuyển hóa (pH> 7,45, HCO3- > 26) là quá trình gây tăng trực tiếp nồng độ HCO3- huyết tương.
- Được sinh ra do mất H+ hoặc nhận lượng lớn HCO3-.
- Được duy trì nhờ sự giữ HCO3- bất thường của thận (thường do suy thận).
Nguyên nhân nhiễm kiềm chuyển hóa thường liên quan đến hạ K máu.
+ Hội chứng giảm thể tích dịch ngoại bào: Thận sẽ tăng giữ HCO3- khi có giảm thể tích dịch ngoại bào (bằng tái hấp thu chung với Na+ ở ống lượng gần), kể cả khi HCO3- huyết tương cao. Một số trường hợp cụ thể thường gặp:
- Nôn mửa.
- Hút dịch vị: mất HCl gây tăng trực tiếp HCO3- huyết tương vì mất một ion H+ tương đương thêm một ion HCO3-.
- Tiêu chảy mạn.
- Ngộ độc thuốc nhuận tràng.
+ Hội chứng thừa mineralcorticoid:
- Cường aldosterone (nguyên phát hoặc thứ phát sau các bệnh mạch máu thận, tăng huyết áp ác tính, …).
- Hội chứng Cushing.
- Tiết ACTH lạc chỗ.
+ Hội chứng Gitelman.
+ Hội chứng Bartter.
4/ Nhiễm toan chuyển hóa:
Nhiễm toan chuyển hóa (pH< 7,35, HCO3- <22) là quá trình gây ra giảm trực tiếp nồng độ HCO3- huyết tương, được sinh ra từ nhận một lượng H+ hoặc mất HCO3-.
+ Nhận H+ từ:
- Tăng sản xuất H+ nội sinh: nhiễm toan ketone, nhiễm toan lactic, ngộ độc salycylate (aspirin). Sulfuric acid hay phosphoric acid có nguồn gốc từ chuyển hóa đạm, thể ketone (ketone body) như acetoacetic acid, beta-hydroxybutyric acid có nguồn gốc từ chuyển hóa lipid còn lactic acid có nguồn gốc từ chuyển hóa đường.
- Sản phẩm chuyển hóa các chất độc qua đường ăn uống như Methanol. Ethylene Glycol và Paraldehyde.
- Giảm sự bài xuất ion H+ của thận, như trong nhiễm toan urea và nhiễm toan ống thận xa type 1.
+ Mất HCO3- qua:
- Mất qua thận, như trong nhiễm toan ống thận gần type II.
- Mất qua tiêu hóa, như trong tiêu chảy.
Ta thường phân loại nhiễm toan chuyển hóa theo khoảng gap anion (AG).
Trước hết, AG là gì?
Khoảng gap anion chính là hiệu số giữa lượng anion không định lượng được và lượng cation không định lượng được. Ta biết trong cơ thể có sự cân bằng giữa các cation và anion. Cation trong cơ thể bao gồm 2 cation chính là Na+ và K+, ngoài ra còn có một số cation không định lượng được (protein mang điện tích dương, Ca2+, Mg2+, …). Anion trong cơ thể bao gồm 2 anion chính là Cl- và HCO3-, ngoài ra còn có một số anion không định lượng được (protein mang điện tích âm. Như vậy:
(Na+) + (K+) + Cation không định lượng = (Cl-) + (HCO3-) + Anion không định lượng.
Do đó, AG = Anion không định lượng – Cation không định lượng = (Na+) + (K+) – (Cl-) – (HCO3-). Để đơn giản, người ta thường lấy AG= (Na+)- (Cl-) – (HCO3-). Giá trị này ở người bình thường nằm trong khoảng 8-12 mmol/l.
- Nhiễm toan chuyển hóa có tăng AG:
Dựa trên công thức, ta dễ nghĩ ngay nguyên nhân làm tăng AG chính là do xuất hiện anion “lạ” làm tăng lượng anion không định lượng (lượng cation không định lượng rất hiếm khi giảm nên không xét ở đây). Nguyên nhân là do tăng sản xuất acid mới/ ngộ độc acid mới:
Chẳng hạn cơ thể bệnh nhân xuất hiện một acid mới (HA). Acid này sẽ được phân ly thành H+ và A-. Ion H+ kết hợp với HCO3- tạo H2O và CO2 và được đào thải ra ngoài => HCO3- giảm. Để đảm bảo cân bằng điện tích, ion A- sẽ đóng vai trò thay thế lượng HCO3- giảm đi, nhưng A- được xếp là 1 anion không định lượng được. Điều này có nghĩa ta đã thay thế 1 anion định lượng (HCO3-) được bởi 1 anion không định lượng được (A-) => AG tăng. Cơ chế này khác với cơ chế ở nhiễm toan chuyển hóa không có tăng AG (sẽ nói rõ hơn ở phần dưới).
| Tăng sản xuất acid hữu cơ | Ngộ độc một chất có tính acid |
| Tăng lactic acid | Ngộ độc methanol |
| Nhiễm toan ketone đái tháo đường | Ngộ độc ethylene glycol |
Lưu ý rằng AG tăng từ 30 trở lên gợi ý một nhiễm toan tăng AG dù pH có thể bình thường.
- Nhiễm toan chuyển hóa không tăng AG:
Nguyên nhân chỉ đơn thuần là do giảm HCO3-/ giảm bài tiết H+ qua thận (do tăng 1 H+ tương đương giảm 1 HCO3-):
Khi HCO3- giảm, để bù trừ, do không có ion A- như ở trường hợp trên, Cl- sẽ phải tăng lên nhằm đảm bảo lượng anion không đổi. Khi đó tổng (Cl-) + (HCO3-) không đổi nên theo công thức ở trên, AG bình thường. Thường gặp trong:
| Giảm HCO3- | Giảm bài tiết H+ qua thận |
| Tiêu chảy | Suy thận |
| Toan hóa ống thận type 2 | Toan hóa ống thận type 1 |
| Acetazoamide (thuốc lợi tiểu) | Toan hóa ống thận type 4 |
Do tình trạng này đi kèm tăng Cl- (để bù trừ) nên người ta cũng gọi nhiễm toan chuyển hóa không tăng AG là nhiễm toan chuyển hóa tăng Cl-.
IV. Cách tính sự bù trừ hợp lí trong rối loạn toan kiềm:
| Rối loạn | Bù trừ của cơ thể |
| Toan chuyển hóa | Công thức Winter: PaCO2 = 1,5. (HCO3-) + 8 (±) 2 |
| Kiềm chuyển hóa | PaCO2 = 40 + 0,7. [(HCO3-) – 24] (±) 5 |
| Toan hô hấp | Cấp tính:
HCO3- = 24 + (PaCO2 – 40)/10 (±) 3 pH = 7,4 + 0,008. (PaCO2 – 40) |
| Mạn tính:
HCO3- = 24 + 4. (PaCO2 – 40)/10 (±) 3 pH = 7,4 + 0,003. (PaCO2 – 40) |
|
| Kiềm hô hấp | Cấp tính:
HCO3- = 24 – 2. (40 – PaCO2)/10 (±) 3 pH = 7,4 – 0,008. (40 – PaCO2) |
| Mạn tính:
HCO3- = 24 – 5. (40 – PaCO2)/10 (±) 3 pH = 7,4 – 0,017. (40 – PaCO2) |
V. Một số đẳng thức ước đoán có giá trị và biện luận một số trường hợp phối hợp:
1/ Quan hệ HCO3- và AG:
Dựa vào công thức tính AG, ta kỳ vọng rằng nếu AG tăng trong nhiễm toan chuyển hóa tăng AG thì HCO3- dự kiến sẽ giảm một khoảng tương đương.
Tuy vậy, mối quan hệ 1:1 này thường ít khi xảy ra. Ta nhớ lại toan chuyển hóa tăng AG là do xuất hiện anion “lạ” có nguồn gốc từ một acid HA (có nguồn gốc nội sinh hoặc ngoại sinh). Sự đệm H+ từ acid này, ngoài HCO3-, còn nhờ nhiều hệ đệm khác. Đó là lí do HCO3- thường giảm một lượng ít hơn sự tăng AG.
Nhiễm toan lactic và nhiễm toan ceton thường có thể dự đoán theo mối tương quan này. Lưu ý D viết tắt cho Delta (độ chênh).
- Nhiễm toan lactic:
DAG/ DHCO3- = 1,5
- Nhiễm toan ceton:
DAG/ DHCO3- = 1
(một số anion ceton làm tăng AG bị mất theo nước tiểu)
Hệ quả:
- HCO3- định lượng cao hơn nhiều so với giá trị dự đoán tương quan với AG tăng gợi ý sự có mặt nhiễm kiềm chuyển hóa “ẩn giấu”.
- HCO3- định lượng thấp hơn nhiều so với giá trị dự đoán tương quan với AG tăng gợi ý sự có mặt của nhiễm toan chuyển hóa không tăng AG “ẩn giấu”.
2/ Quan hệ giữa pH và PaCO2:
Theo các công thức bù trừ ở mục IV, ta để ý rằng trong rối loạn hô hấp cấp, DpH thường xấp xỉ 0,008 lần DCO2. Đây là một chỉ điểm để đánh giá rối loạn hô hấp của bệnh nhân lúc nhập viện là cấp hay mạn.
3/ HCO3- nằm giữa 2 giá trị dự đoán:
Nếu gặp một bệnh nhân toan hô hấp và có giá trị HCO3- định lượng nằm giữa 2 giá trị dự đoán (1 giá trị bù trừ cho trường hợp toan hô hấp cấp và 1 giá trị bù trừ cho toan hô hấp mạn). 3 trường hợp có thể xảy ra:
- Toan chuyển hóa xuất hiện trên một toan hô hấp mạn.
- Toan hô hấp cấp xuất hiện trên một toan hô hấp mạn.
- Toan hô hấp cấp đi kèm kiềm chuyển hóa.
Ngược lại, nếu bệnh nhân kiềm hô hấp và có giá trị HCO3- như vậy, cũng có 3 trường hợp xảy ra:
- Kiềm chuyển hóa xuất hiện trên một kiềm hô hấp mạn.
- Kiềm hô hấp cấp xuất hiện trên một kiềm hô hấp mạn.
- Kiềm hô hấp cấp đi kèm toan chuyển hóa.
Biện luận các trường hợp này cần dựa vào bệnh cảnh lâm sàng để loại trừ.
* BẠN CÓ BIẾT:
1/ Hb có thể đệm cho cân bằng toan- kiềm như thế nào?
Trong hồng cầu có men anhydrase carbonic. Men này xúc tác phản ứng thuận nghịch CO2+ H2O tạo H2CO3. Nhờ phản ứng này mà Hb có thể nhả hoặc nhận CO2 từ mô (hình dưới), qua đó điều chỉnh pH mô. Xem thêm https://nguyenhuythekopites.wordpress.com/2018/11/22/doc-ket-qua-khi-mau-dong-mach/.

Source: https://www.youtube.com/watch?v=W7egri8t-iE
2/ Hội chứng Guillain- Barré là gì?
Đây là một rối loạn tự miễn khá hiếm gặp, thường do một đợt nhiễm trùng. Hệ miễn dịch cơ thể tấn công các dây thần kinh, đặc biệt là hệ thần kinh ngoại biên. Biến chứng nguy hiểm nhất là khi thần kinh chi phối các cơ hô hấp hoặc thần kinh tự chủ bị ảnh hưởng, khiến bệnh nhân suy hô hấp hoặc rối loạn thần kinh tự chủ.

Source: Mayo Clinic Network
3/ Tại sao nguyên nhân nhiễm kiềm chuyển hóa thường liên quan đến hạ K máu?
Cơ chế của hiện tượng này tương đối phức tạp. Một số giả thiết được đặt ra bao gồm tình trạng hạ K máu do mất dịch (nôn mửa, tiêu chảy, …) hoặc là hậu quả thứ phát của sự tăng tiết aldosterone (tái hấp thu Na+ và H2O, thải K+).
Mở rộng một số nguyên nhân gây tăng K+ máu hoặc hạ K+ máu.
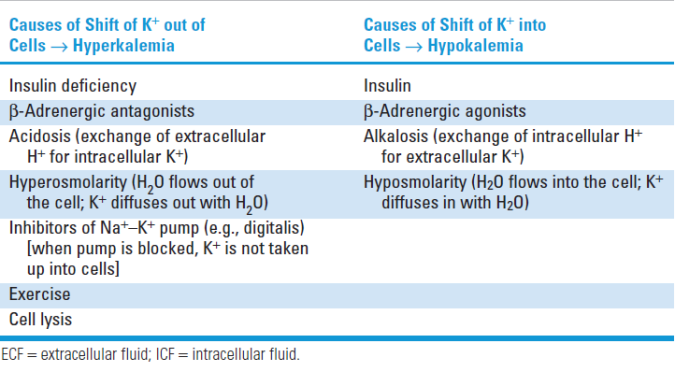
Source: BRS Physiology
4/ Hội chứng Bartter là gì? Hội chứng Gitelman là gì?
Cả 2 đều liên quan đến khiếm khuyết tái hấp thu Na+ – Cl- ở thận, tuy nhiên có một số điểm cần phân biệt. Nồng độ Ca2+ niệu cao ở hội chứng Bartter và không tăng ở hội chứng Gitelman là tiêu chuẩn thường dùng để phân biệt 2 hội chứng này.

Source: Saudi Journal of Kidney Disease and Transplantation
5/ Glucocorticoid (như cortisol) với mineralcorticoid (như aldosterone) có gì khác nhau?
Cả 2 đều là các corticosteroid hormone, cùng được tiết từ vỏ thượng thận nhưng glucocorticoid (như cortisol/hydrocortisone) liên quan đến chuyển hóa đường, trong khi mineralcorticoid (như aldosterone) chịu trách nhiệm trong chuyển hóa muối khoáng (như các chất điện giải Na+, K+, …).

Source: http://fafunderecsemb.cf/pogo/is-prednisone-a-glucocorticoid-or-mineralocorticoid-xuhu.php
Chúng cũng khác nhau về “xuất xứ” trong vỏ thượng thận: glucocorticoid được tiết từ lớp cầu/ lớp ngoài (zona glomerulosa) trong khi mineralcortcoid có nguồn gốc từ lớp sợi/ lớp giữa (zona fasciculata). Lớp còn lại của vỏ thượng thận, tức lớp lưới/ lớp trong (zona reticularis) tiết hormone steroid sinh dục (sex steroid, ví dụ androgen).

Source: https://www.dartmouth.edu/~anatomy/Histo/lab_6/endocrine/DMS164/popup.html
6/ Các loại toan hóa ống thận (renal tubular acidosis- RTA)?
Trước hết, nhắc lại sinh lý thận liên quan đến bài tiết H+, tái hấp thu HCO3- và tiết ammonia (NH3):
- H+ được bài tiết (excretion) ở ống lượn xa.
- 90% HCO3- được tái hấp thu (reabsorption) ở ống lượn gần, bình thường không có HCO3- trong nước tiểu.
- NH3 được tiết (secretion) để kết hợp với H+ ở ống lượn xa, tạo NH4+ (amonium), góp phần thải H+ ra ngoài.
Toan hóa ống thận là tình trạng thận không thể làm toan nước tiểu như bình thường, dẫn đến H+ tích tụ trong cơ thể. Các type toan hóa ống thận dưới đây đều gây nhiễm toan chuyển hóa không tăng AG.
Chỉ số AG niệu, được tính bằng (Na+)+ (K+) – (Cl-), bình thường nằm trong đoạn [-20;0] và có thể được dùng để phân biệt các type. Lưu ý rằng tính âm/ dương của AG niệu được quyết định phần lớn do Cl-.
+ Type 1: toan hóa ống thận xa (distal RTA), do mất khả năng bài tiết H+ dưới dạng NH4+ ở ống lượn xa hoặc ống góp.
Về mặt phân tử, type 1 được đặc trưng bởi sự kém hiệu quả của kênh H+/K+ – ATPase ở các tế bào alpha xen giữa của ống góp (alpha – intercalated cell). Kênh này đóng vai trò đẩy H+ ra lòng ống (lumen) để thải ra ngoài, đồng thời tái hấp thu K+ vào biểu mô rồi trở lại máu. Tế bào alpha xen giữa của ống góp có khả năng bài tiết H+ theo cơ chế tích cực nguyên phát (primary active transport), chống lại bậc thang nồng độ (concentration gradient) cao gấp 1000 lần, trong khi sự bài tiết tích cực thứ phát ở ống gần hay các phần khác của ống thận chỉ chống lại được bậc thang nồng độ cao gấp vài ba lần. Đó là lí do tình trạng toan ống thận type 1 được xem là trầm trọng nhất, và chỉ ở trường hợp này mới có pH niệu > 5,5.
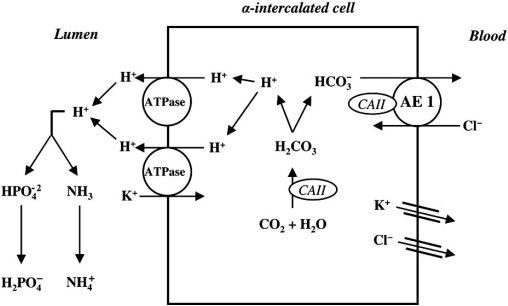
Source: https://openi.nlm.nih.gov/detailedresult.php?img=PMC2699831_CG-10-51_F2&req=4
Hoạt động kênh này kém đi sẽ dẫn đến H+ tích tụ trong cơ thể (không được thải qua nước tiểu như bình thường), đồng thời việc tái hấp thu K+ cũng giảm đi. Do đó, pH niệu tăng, K+ huyết tương giảm. H+ giảm trong nước tiểu kéo theo Cl- niệu giảm theo (cân bằng điện tích) => AG niệu > 0.
Tình trạng toan máu này có thể kéo theo tăng Ca2+ niệu. Nguyên nhân có thể do xương, vốn giàu Ca2+ và phosphate, phải giải phóng phosphate để đệm pH, kéo theo Ca2+ ra ngoài và được thải qua nước tiểu. Điều này làm tăng khả năng tạo sỏi, đặc biệt là sỏi Calcium Phosphate. Sỏi Calcium Phosphate càng được tăng cường ở pH niệu cao (phù hợp với pH > 5,5 như trên). Nguyên nhân thường gặp của RTA type 1 bao gồm các nguyên nhân phá hủy tế bào alpha xen giữa của ống góp (như lạm dụng thuốc kháng nấm Amphoteracin B hay tắc niệu đạo- obstructive uropathy) và hội chứng tự miễn Sjogren khi các tế bào miễn dịch tấn công kênh H+/K+ – ATPase.
+ Type 2: toan hóa ống lượn gần (proximal RTA), do suy giảm tái hấp thu HCO3- ở ống lượn gần.

Source: https://www.anaesthesiamcq.com/AcidBaseBook/ab2_4.php
Sơ đồ trên chỉ ra ở điều kiện bình thường, kênh đối chuyển Na+/ H+ và kênh đồng chuyển Na+/ HCO3- ở tế bào ống lượn gần (Proximal Convoluted Tubular Cell – PCT) giúp thải chủ động vào lòng ống (PCT lumen) 1 ion H+, đồng thời tái hấp thu vào mao mạch quanh ống (Peritubular capillary) 1 ion HCO3-, qua 3 giai đoạn chính:
- Vận chuyển chủ động Na+ ra khoảng kẽ nhờ kênh Na+/K+ – ATPase.
- Na+ được tái hấp thu vào biểu mô ống thận nhờ gradient được tạo ra ở giai đoạn 1. Quá trình này đồng thời thải tích cực ion H+ ra nước tiểu. H+ cũng được bài tiết chủ động qua kênh H+ – ATPase.
- Ion H+ này kết hợp với ion HCO3-, trải qua nhiều quá trình biến đổi nhờ sự xúc tác của men carbonic anhydrase để đưa HCO3- vào lại biểu mô ống thận (thông qua dạng trung gian là CO2 hòa tan cao trong lớp lipid của màng) và được tái hấp thu bởi Na+ qua kênh đồng chuyển Na+/HCO3-.
RTA type 2 xảy ra do bất thường ở ít nhất 1 trong 3 bước trên, dẫn đến kém tái hấp thu HCO3-.
Sự tái hấp thu HCO3- kém đi đồng nghĩa nhiều HCO3- được thải thụ động qua nước tiểu (không thể kết hợp H+ để được tái hấp thu). Nồng độ HCO3- niệu cao làm ức chế hoạt động đối chuyển HCO3-/Cl- (đưa HCO3- vào nước tiểu, đưa Cl- trở lại biểu mô ống thận) => Cl- niệu cao => AG niệu âm.
Ngoài ra, do HCO3- được tái hấp thu cùng Na+ nên nếu HCO3- được tái hấp thu kém đi thì Na+ cũng ít được tái hấp thu vào máu hơn => tăng tiết aldosterone từ lớp cầu ở vỏ thượng thận => tăng thải K+ => K+ huyết tương thấp.
Nguyên nhân thường gặp:
- Hội chứng Falconi, gây giảm sút khả năng tái hấp thu ở tế bào ống lượn gần.

Source: http://www.stepwards.com/?page_id=1882
Theo hình trên, hội chứng Falconi làm việc tái hấp thu Na+, PO4-, HCO3-, glucose và amino acid bị giảm sút. Không khó hiểu khi bệnh nhân bị hạ phosphate máu, hạ glucose máu, … bên cạnh RTA type 2.
- Các tổn thương di truyền do các gốc oxi hóa (Hereditary – Oxidative injury):
Các nguyên nhân trong nhóm này đều dẫn đến sự tích tụ bất thường một chất trong các cơ quan trong cơ thể, đặc biệt là thận, gây rối loạn chuyển hóa, tổn thương khả năng tái hấp thu của tế bào ống lượn gần. Chất đó trong Cystinosis là amino acid cystine, trong Tyrosinemia là Tyrosine, trong hội chứng Wilson là đồng, trong bệnh ứ đọng glycogen (Glycogen Storage Disease – GSD) là glycogen, …
- Ứ đọng chuỗi nhẹ immunoglobulin mắc phải (Light chain deposition disease):
Thường gặp trong Amyloidosis (tích tụ protein amyloid vốn thường được sản xuất từ tủy xương), đa u tủy xương (multiple myeloma).
- Thuốc ức chế Carbonic Anhydrase.
+ Type 3: không còn được xem là 1 type riêng biệt.
+ Type 4: toan hóa tăng K+ máu ở ống lượn xa (hyperkalemic RTA), gây nhiễm toan chuyển hóa không tăng anion gap. Nguyên nhân là do thiếu/ kháng aldosterone. AG niệu > 0.

Source: https://medcomic.com/medcomic/renal-tubular-acidosis/
Tóm tắt đặc điểm các type 1, 2, 4:
| Type 1 RTA | Type 2 RTA | Type 4 RTA | |
| Bất thường | Bất thường bài tiết H+/ tiết NH4+ | Bất thường tái hấp thu HCO3- | Thiếu/ kháng aldosterone |
| Vị trí có bất thường | Ống lượn xa/ ống góp | Ống lượn gần | Ống lượn xa |
| pH niệu | < 5,5 | > 5,5 | > 5,5 |
| AG niệu | > 0 | < -20 | > 0 |
| HCO3- huyết tương | < 15 | 15-18 | > 18 |
| K+ huyết tương | Thấp | Thấp | Cao |
| Calcium niệu | Cao | — | — |
| Khả năng tạo sỏi | Cao | Hiếm | Hiếm |
| Citrate niệu | Thấp | Cao | Thấp |
Lưu ý giá trị AG niệu < -20 còn gặp trong toan chuyển hóa do tiêu chảy. Nói ngắn gọn, bệnh nhân toan chuyển hóa có AG niệu < -20 gợi ý mất HCO3- qua tiêu chảy hoặc toan hóa ống lượn gần, AG niệu >0 gợi ý toan hóa ống lượn xa.
7/ Thuốc lợi tiểu Acetazoamide (nay thường được dùng để hạ nhãn áp) gây giảm HCO3- huyết tương bằng cách nào?

Source: https://ejhp.bmj.com/content/22/6/328
Acetazoamide ức chế men carbonic anhydrase, từ đó làm giảm tổng hợp H+ trong tế bào biểu mô ống thận, kéo theo giảm hoạt động kênh đối chuyển Na+/ H+ (Kênh đẩy Na+ vào trong tế bào còn H+ ra nước tiểu) khiến cho Na+ và HCO3- bị “bẫy” trong nước tiểu ra ngoài => giảm tái hấp thu HCO3- => giảm HCO3- huyết tương.
8/ Suy thận chịu trách nhiệm trong cả nhiễm toan và nhiễm kiềm chuyển hóa?
Suy thận có khả năng gây nhiễm toan chuyển hóa có AG bình thường (suy khả năng tạo ammonia ở thận nên giảm khả năng đào thải H+). Trong nhiễm kiềm chuyển hóa, suy thận đóng vai trò duy trì do giảm khả năng thải HCO3-.
9/ Albumin là 1 anion không định lượng được. Vậy ở bệnh nhân giảm albumin máu (như viêm gan) thì cần tính AG như thế nào để tránh nhiễu?
Albumin bị giảm 1g/dl sẽ làm giảm 2,5 AG. Với giá trị albumin chuẩn là 4g/dl thì có thể chuyển AG bệnh nhân bị giảm albumin máu về tình trạng không giảm albumin máu.
AG điều chỉnh = AG do giảm albumin máu + (4 – Albumin) x 2,5
Ví dụ một bệnh nhân viêm gan có albumin huyết tương là 1,5g/dl, có AG là 14. Như vậy nếu bệnh nhân không bị giảm albumin huyết tương thì AG điều chỉnh = 14 + (4 – 1,5) x 2,5 = 20.

